GROMACS¶
GROMACS (GROningen MAchine for Chemical Simulations) is a package to perform molecular dynamics simulations, i.e. simulate the Newtonian equations of motion for systems with hundreds to millions of particles. The following description is from the GROMACS Home page: http://www.gromacs.org. – “It is primarily designed for biochemical molecules like proteins, lipids and nucleic acids that have a lot of complicated bonded interactions, but since GROMACS is extremely fast at calculating the nonbonded interactions (that usually dominate simulations) many groups are also using it for research on non-biological systems, e.g. polymers.”
Recent versions of this has been compiled under Centos 8 and
can be found under /shared/opt/.
You can also compile your own version in your home directory.
- /shared/opt/gromacs-2018.8
- /shared/opt/gromacs-2019.6
- /shared/opt/gromacs-2020.4
- /shared/opt/gromacs-2021.4
Thanks to Sheikh Imamul Hossain for proving some of the data and usage information about GROMACS on this page.
Running GROMACS¶
Read the GROMACS documentation. There are several excellent tutorials by Justin Lemkul here GROMACS Tutorials.
We assume you will be using a GROMACS version under /shared/opt/.
Setup GROMAC’s environment:
$ source /shared/opt/gromacs-2021.4/bin/GMXRC.bash
The following commands should give you a summary of the build information and a help page for the mdrun command:
$ gmx --version
$ gmx mdrun -h
Of course GROMACS runs should be done via a PBS submission script. Below is an example. Ypou will need to work out suitable values for the number of CPUs, memory and walltime for your own simulations.
#!/bin/bash
#PBS -N GROMACS
#PBS -l walltime=0:30:00
#PBS -l ncpus=16
#PBS -l mem=8GB
#PBS -m abe
#PBS -M Your.Email@uts.edu.au
# Note: sourcing GMXRC.bash will start a new shell which will change your directory to $HOME.
# Therefore make sure that the "cd $PBS_O_WORKDIR" comes after the source command. This will
# change your working directory back to the PBS job directory.
source /shared/opt/gromacs-2021.4/bin/GMXRC.bash
cd $PBS_O_WORKDIR
gmx mdrun -nt $NCPUS -pin on -deffnm npt
How Many Cores do I Need?¶
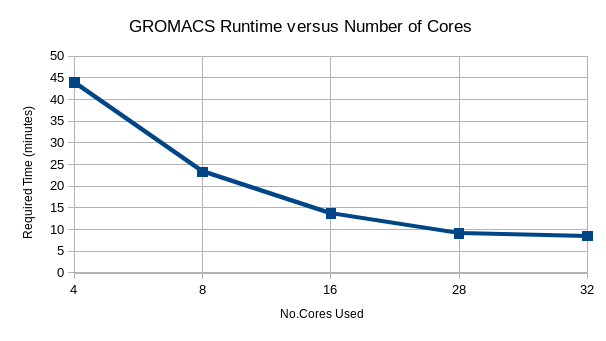
Just asking for more cores will not necessarily give you better performance. Generally for applications that are mult-threaded you will get diminishing speedups as the number of cores requested increases, as can be seen in the real world example for a GROMACS job below. For this user they decided to use 28 cores as there was no significant speedup using any more cores. Requesting less cores also means they have a better chance of their job running sooner when the cluster is very busy.
You should test your applications ability to scale with data sets representative of your work. Perform runs with more cores and note the time required.
Problems¶
Note that some GROMACS versions (maybe all) have a bug;
gmx writes normal output to stderr - Redmine #2211.
This means that when you run GROMACS production runs under PBS (which you always need to do)
the PBS standard output file (.o<job_id>) will be empty and your PBS standard error file
(.e<job_id>) will contain your standard output!